Glomuvenous malformations
Published Web Location
https://doi.org/10.5070/D37xm7h2ccMain Content
Glomuvenous malformations
J Scott Henning DO, Olympia I Kovich MD, Julie V Schaffer MD
Dermatology Online Journal 13 (1): 17
New York University Department of DermatologyAbstract
A 9-year-old girl presented with a congenital, blue-purple, partially compressible plaque with a cobblestone surface on the left lateral foot and ankle. Similar, solitary, blue nodules later appeared elsewhere on the extremities. The lesions were tender to palpation and were associated with spontaneous paroxysms of pain and paresthesias. Histopathologic evaluation of a skin biopsy specimen showed rows of glomus cells that surrounded thin-walled vascular channels, which confirmed the diagnosis of glomuvenous malformations. This autosomal dominant condition, which is due to mutations in the GLMN gene, presents with clinical findings that are distinct from those of familial, multiple, cutaneous and mucosal venous malformations. Treatment options include excision, sclerotherapy, and laser therapy (ablative or pulsed dye).
Clinical synopsis
A 9-year-old girl presented to the Charles C. Harris Skin and Cancer Pavilion Pediatric Dermatology Section in February, 2005, for the evaluation of a congenital vascular anomaly on the left lower leg and foot. Over time, the lesion had become thicker and increased in diameter in proportion to the patient's growth. She reported a 1-year history of daily paroxysms of pain localized to the affected area; these episodes usually lasted several minutes and were occasionally precipitated by changes in temperature or pressure. In 2005, several, tender, blue nodules also appeared elsewhere on her upper and lower extremities. The patient had no other medical problems and denied abdominal pain or hematochezia. There was no family history of similar lesions. After confirmation of the diagnosis by a skin biopsy, a trial of sclerotherapy with sodium tetradecyl sulfate to the affected superficial vessels of the left foot led to slight flattening of the lesions but no pain relief. In the past several months, the patient has noted intermittent, seemingly spontaneous paresthesias of the upper and lower extremities in areas without clinical evidence of vascular lesions.
Multiple, 0.5-1.0 cm, blue-purple, partially compressible nodules coalesced to form a 15- x 4-cm plaque with a cobblestone surface on the left lateral foot and ankle. Solitary, blue nodules also were present on the right posterior thigh, right lateral calf, and right upper arm. All of the lesions were tender to palpation. Prominent veins were noted on the left calf and popliteal fossa, but the legs were symmetric in circumference and length.
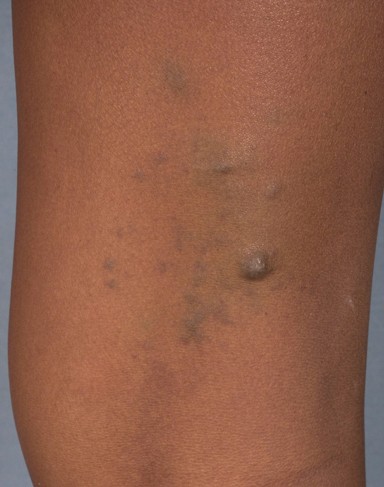 | 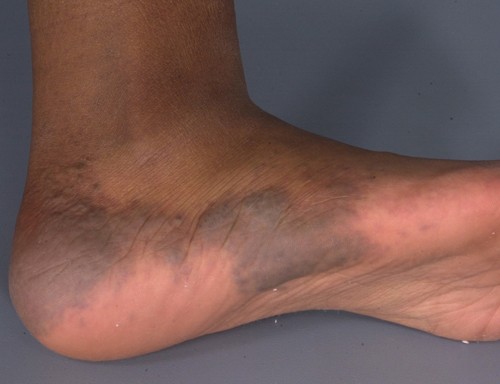 |
| Figure 1 | Figure 2 |
|---|---|
A magnetic resonance image and magnetic resonance angiography of the right foot showed a vascular malformation in the lateral right hindfoot, midfoot, and calf that was compatible with a venous malformation. Angiographic sequence was unremarkable, with normal three-vessel runoff through the calf to the foot and a normal deep venous system.
 |
| Figure 3 |
|---|
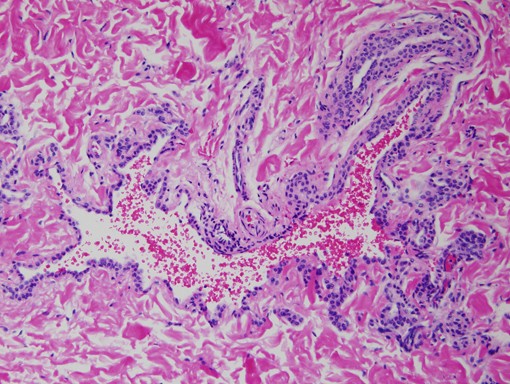
Histopathology reveals a large, dilated blood vessel lined by thin endothelial cells, a thin layer of smooth muscle fibers, and several rows of glomus cells.
Comment
Glomuvenous malformations (GVMs; previously referred to as glomangiomas or glomus tumors) are a subtype of venous malformations (VM) that are characterized by rows of glomus cells that surround distorted, thin-walled vascular channels. The glomus cells closely resemble the modified smooth muscle cells of a specialized arteriovenous anastamosis within the dermis, the Sucquet-Hoyer canal or glomus body, which represents a neuromyoarterial receptor that is involved in temperature regulation [1]. Sporadic, solitary GVMs most commonly arise in the nail bed, which is a site with a high density of glomus bodies.
Multiple GVMs occurring in a widespread or segmental distribution can be inherited in an autosomal dominant pattern with incomplete penetrance (90 % by age 20 years) [3]. Affected individuals and carriers have heterozygous germline mutations in the GLMN gene that cause a premature stop codon and a truncated glomulin protein [3, 4]. There is evidence that the GVMs themselves result from a somatic second hit to GLMN that leads to loss of heterozygosity and localized complete absence of glomulin function (type 2 mosaicism) [3]. Approximately two-thirds of patients with GVMs have a family history of similar lesions as compared to 1 percent of individuals with other types of VMs [2]. The clinical as well as histopathologic features of GVMs are distinct from those of multiple cutaneous and mucosal VMs, which is a familial form of classic VMs (see Table) [2].
The treatment of choice for isolated cutaneous GVMs is surgical excision. However, this may be impractical in cases of multiple or large segmental lesions. Sclerotherapy with sodium tetradecyl sulfate, polidocanol, and hypertonic saline has been reported to be effective in patients with multiple GVMs that are located on the extremities, whereas the use of sclerosants including polidocanol, pure ethanol, and Ethibloc® (a mixture of zein, sodium amidotrizoate tetrahydrate, oleum papaveris, and propylene glycol) was unsuccessful in a series of seven patients with large facial GVMS [5, 6]. Ablative therapy with argon and carbon dioxide lasers is of potential benefit for small, superficial lesions. Treatment with the pulsed dye laser may also help to flatten GVMs and provide pain relief [7]. Magnetic resonance imaging can be useful in defining the extent of lesions and their relationship to other anatomic structures prior to therapeutic interventions [6].
References
1. Blume-Peytavi U, et al. Multiple familial cutaneous glomangioma: a pedigree of 4 generations and critical analysis of histologic and genetic differences of glomus tumors. J Am Acad Dermatol 2000;42:6332. Boon LM, et al. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol 2004;140:971
3. Brouillard P, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations ("glomangiomas"). Am J Hum Genet 2002;70:866
4. Brouillard P, et al. Four common glomulin mutations cause two thirds of glomuvenous malformations ("familial glomangiomas"): evidence for a founder effect. J Med Genet 2005;42:e13
5. Parsi K, Kossard S. Multiple hereditary glomangiomas: Successful treatment with sclerotherapy. Australas J Dermatol 2002;43:43
6. Mounayer C, et al. Facial glomangiomas: large facial venous malformations with glomus cells. J Am Acad Dermatol 2001;45:239
7. Antony FC, et al. Complete pain relief following treatment of a glomangiomyoma with the pulsed dye laser. Clin Exp Dermatol 2003;28:617
© 2007 Dermatology Online Journal