Familial multiple angiolipomatosis
Published Web Location
https://doi.org/10.5070/D36pj611k2Main Content
Familial multiple angiolipomatosis
Naheed R Abbasi MD, Isaac Brownell MD PhD, William Fangman MD
Dermatology Online Journal 13 (1): 3
New York University Department of DermatologyAbstract
An 80-year-old man presented with a 50-year history of asymptomatic, subcutaneous masses on the arms, trunk, and legs. His father and maternal grandmother had had similar lesions. Histopathologic examination showed a benign angiolipoma; the same diagnosis has been made on several previous biopsy specimens. This patient's history and physical examination support the diagnosis of familial angiolipomatosis, which is a benign, autosomal-dominant condition that may be regarded as a subtype of familial multiple lipomatosis (FML) or as a distinct entity. Management of this condition may include liposuction or surgery to reduce the tumor burden.
Clinical synopsis
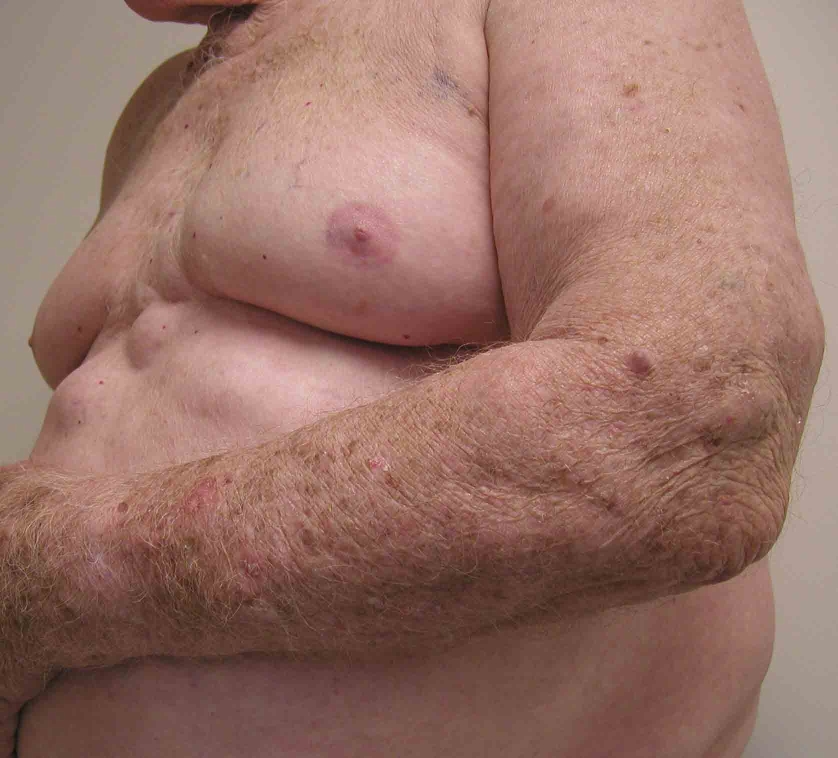
An 80-year-old man presented to the Veterans Affairs New York Harbor View Healthcare System Dermatology Service in September 2005 with a 50-year history of subcutaneous masses on the abdomen, upper arms, and right thigh. The patient stated that these lumpy masses, while neither painful nor pruritic, have caused cosmetic concern. Both his father and paternal grandmother had a history of similar subcutaneous masses for which they had not sought treatment. A dermatologist had excised a few of the subcutaneous masses and reassured the patient that they were not malignant. There were no joint symptoms.
The patient also has a history of coronary artery disease, hyperlipidemia, multiple myeloma, and prostate cancer. The onset of these conditions occurred after the development of the subcutaneous masses by years to decades. The subcutaneous masses were not associated with antecedent medications or systemic infections. Medications included simvastatin, aspirin, atenolol, and razosin.
Numerous, 1-10-cm, firm, rubbery, subcutaneous masses were present on the upper arms, anterior portions of the abdomen, and right thigh. Some lesions were clustered and others were solitary, but each was encapsulated. There were no visible changes of the overlying skin and no tan macules. No mucosal abnormalities were noted. There was no axillary, inguinal, or other lymphadenopathy.
 |  |
| Figure 1 | Figure 2 |
|---|---|
A complete blood count, chemistry panel, hepatic function panel, total cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides were normal.
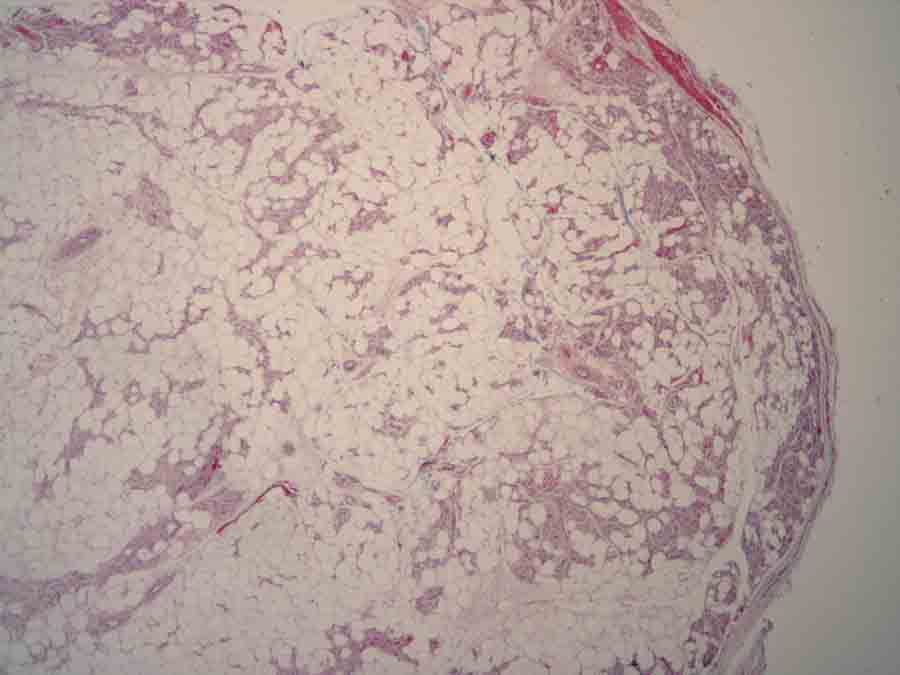
Histopathology reveals a circumscribed neoplasm composed of mature adipocytes and increased numbers of thin-walled blood vessels. Scattered fibrin thrombi are noted within many of the vascular lumens.
Comment
Familial multiple lipomatosis (FML) is a rare entity that is characterized by numerous, encapsulated lipomas on the trunk and extremities. It generally affects consecutive generations within families. No accurate assessment of the prevalence of this condition is available, and various authors have found a slightly increased predilection in men [1, 2]. Familial multiple lipomatosis must be differentiated from multiple symmetric lipomatosis (MSL), which is a condition of diffuse and symmetric fatty infiltration that occurrs in middle-aged men and those with a history of alcoholism [3]. A distinction between the two conditions is the relative sparing of the neck and shoulders in FML and the presence of discrete lipomas rather than the diffuse and symmetric lipomatous infiltration in MSL [1, 3].
Clinical features of FML include multiple, well-encapsulated, oval-to-round, subcutaneous, rubbery lipomas that range from a few millimeters to 25 centimeters in size [1]. These lipomas may be misdiagnosed as neurofibromas [4]. Lipomas generally first appear in the third through fifth decades of life and occur most commonly on the trunk and extremities [2]. The forearms are a site of frequent occurrence [1]. The bulk of available evidence supports an autosomal dominant mode of inheritance for FML; this hypothesis is supported by high levels of penetrance among first-degree relatives [2, 5].
Familial angiolipomatosis has likely been included within the broader heading of FML since FML was first described in 1846. Only a few of the more recent papers describe the existence of familial angiolipomatosis as a distinct entity [4, 6, 7, 8]. Like FML, familial angiolipomatosis has demonstrated autosomal dominant inheritance [4, 6]. A single paper has described the existence of angiolipomas infiltrating adjacent muscle tissue in two members of the same family [9]. The traditional belief that angiolipomas may be differentiated from lipomas clinically by subjective reports of tenderness or pain in the former [10] is challenged by the case presented here, in which the described patient has undergone numerous excisions of asymptomatic angiolipomas.
Histopathologic evaluation of tissue from patients with hereditary angiolipomatosis shows numerous, variably sized but typical-appearing angiolipomas. Angiolipoma is a subtype of lipoma, in which the vascular component varies from 15 to 50 percent, rather than less than 10 percent as in common lipomas [6].
On a genetic level, FML may result from a translocation of the high-mobility group protein isoform I-C on chromosome 12 and the lipoma preferred partner gene on chromosome 3 [11, 12]. Specific gene rearrangements in familial angiolipomatosis, if distinct, have not been described. Some authors have proposed an association between FML and hyperlipidemia [13].
Familial lipomatosis and angiolipomatosis do not require medical or surgical treatment. Liposuction [8] and surgical excision offer cosmetic and practical benefit to patients whose tumors are disfiguring or unusually large. Surgical techniques have been developed to remove dozens of tumors at once and with a minimal number of incisions and scars [14].
References
1. Leffell DJ, Braverman IM. Familial multiple lipomatosis: report of a case and review of the literature. J Am Acad Dermatol 1986; 15: 2752. Keskin D, et al. Familial multiple lipomatosis. Israel Med Assoc J 2002; 4: 1121
3. Lee MS, et al. Multiple symmetric lipomatosis. J Korean Med Sci 1988; 4: 163
4. Cina SJ, et al. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med 1999; 123: 946
5. Mohar N. Familial multiple lipomatosis. Acta Derm Venereol (Stockh) 1980; 60: 509
6. Kumar R, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet 1989; 35: 202
7. Hapnes SA, et al. Familial angiolipomatosis. Clin Genet 1980; 17: 202
8. Kanter WR, Wolfort FG. Multiple familial angiolipomatosis: treatment of liposuction. Ann Plast Surg 1988; 20: 277
9. Chang JL, et al. Infiltrating angiolipoma: report of a familial occurrence. Orthopedics 1994; 17: 364
10. Osment LS. Cutaneous lipomas and lipomatosis. Surg Gynecol Obstet 1968; 127: 129
11. Toy BR. Familial multiple lipomatosis. Dermatol Online J 2002; 9: 9
12. Mrozek K, et al. Chromosome 12 breakpoints are cytogenetically different in benign and malignant lipogenic tumors. Cancer Res 1993; 53:1670
13. Rubinstein A, et al. Non-symmetric subcutaneous lipomatosis associated with familial combined hyperlipidemia. Br J Dermatol 1989; 120:689
14. Ronan SJ, Broderick T. Minimally invasive approach to familial multiple lipomatosis. Plast Reconstr Surg 2000; 106:878
© 2007 Dermatology Online Journal